
Overcoming The Challenge Of The 2011 FDA Process Validation Guidance

By Eugene Wordehoff, Managing Director, Merton Partners LLC
A paradigm shift is underway in process validation. The FDA revised the guidance to industry for process validation in January 2011. This guidance defines process validation as “the collection and evaluation of data, from the process design stage through commercial production which establishes scientific evidence that a process is capable of consistently delivering quality product.” The new emphasis is on “scientific” rather than “documented” evidence. [1]
This new approach is aimed at increasing process understanding and control. The consequences of ineffective process validation are significant as evidenced by increased cost of compliance: Adverse events, warning letters, delays in product approval and launch, plant remediation, field alerts, recalls and product shortages.
Implications For Industry
Pharmaceutical manufacturers must now make deliberate decisions about which statistical tools and analyses are appropriate for their products and processes. For example, the traditional “rule of three” for the number of product batches required to validate a process has been challenged. The use of statistical tools are now encouraged not only to determine the number of batches required to validate a process but throughout the process life cycle to demonstrate product integrity, safety, and efficacy.
The larger paradigm shift is that the former approach to validation focused only on what is now called Stage 2: Process Qualification. The new guidance looks at the life cycle beginning with Stage 1: Process Design and ending with Stage 3: Continued Process Verification, in addition to the existing Stage 2.
This has produced a vexing problem for many pharmaceutical executives, especially problematic for midsize to small firms. The standards and expectations are being raised at the same time that drug products are becoming more sophisticated. Historical formulations were generally simpler; now products have complex release mechanisms making it more challenging to demonstrate equivalent bioavailability. Risk assessment is now integral to the validation process and must consider supply chain not under direct control, such as global raw material sources. There is also pressure to drive costs down and reduce production cycles.
New questions are raised: How is an understanding of process design demonstrated? What variables must be considered? What are the inputs and outputs? How are the new risk assessments accomplished? How are statistically valid results demonstrated? Is the internal statistical staff current with FDA guidance?
Some of the specific technical statistical skills that would be required to fully cover the new FDA Guidance include Design of Experiments (DOE), sample size determination, linear regression and correlation, nonlinear regression, Analysis of Variance (ANOVA), and measurement system analysis, to name a few. In addition to competency with the statistical tools, the ability to interpret data and integrate the science of the process with the specific statistical tools is critical. The higher the level of physical-chemical knowledge and pharmaceutical science knowledge coupled with a broad base of statistical skills, the better the outcome.
The benefits of statistical analysis are numerous (think “Cheaper/Better/Faster”): accelerated innovation cycle time, reduced total cost of process and product, improved compliance levels, enhanced quality and safety, reduced rework and recalls and improved patient safety and efficacy.
Practical Statistical Applications
The 2011 Guidance promotes a life-cycle approach to process validation that includes scientifically sound design practices, robust qualification, and process verification. The lifecycle concept links product and process development, qualification of the commercial manufacturing process, and maintenance of the process in a state of control during commercial production and throughout the ownership of the product by the manufacturer.
Stage 1- Process Design: The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities. The objective of this stage is to demonstrate understanding and robustness of the design space. Statistical applications include:
- Risk assessment / cause and effect matrix to identify potential variables that impact the process and any risks in the system
- DOE to explore process variables
- Sample size determination to specify the number of samples needed to detect significant differences in the variables
- Multiple regression analysis to determine the significant variables and define the operating regions of the variables
- Development of the control strategy to document the operating ranges for the variables to be controlled
- Analytical method validation to obtain an adequate mechanism for measuring the in-process and release parameters
- Analysis to ensure the measurement system is repeatable and reproducible
Stage 2- Process Qualification: During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing. The objective of this stage is to demonstrate process capability and repeatability. Statistical applications include:
- Sample size determination
- ANOVA to detect inter/intra batch variability
- Development of specifications
- Process capability demonstration
- Refinement of the control strategy to the operator level
- Stability analysis for determination of shelf-life
- Statistical documentation to support to the CMC filing
Stage 3 - Continued Process Verification (CPV): Ongoing assurance is gained during routine production that the process remains in a state of control. The objective of this stage is to demonstrate a stable and effective process. Statistical applications include:
- Customer complaint analysis for ongoing monitoring
- Setting up and ongoing use of control charts
- Ongoing monitoring of process capability
- Implementation of measurements and controls
- Integration of Annual Product Review (APR) statistics and CPV
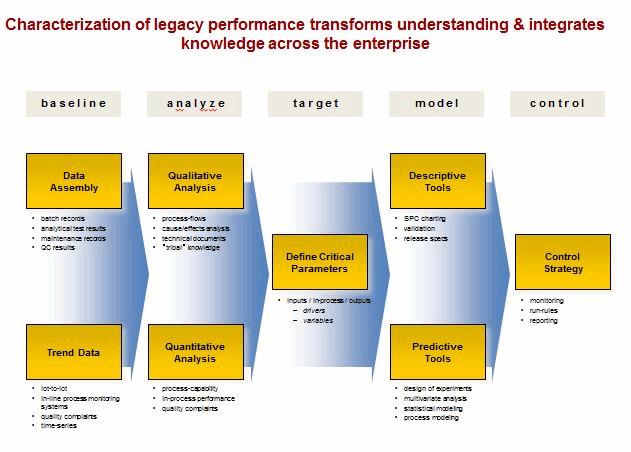
Legacy Products
The three stages of the pharmaceutical life cycle as described above are applied to new products as they are developed and introduced. However, most products currently in operation are legacy products and may not have had the benefit of the new guidance.
Legacy products and processes are vulnerable because knowledge can be fragmented, often caused by: step-wise development cycles, multiple tech transfers, compartmentalized data, incompatible information systems, ineffective supplier relations and aggressive cost reduction. Statistical tools can be used to characterize legacy products and processes in the existing facilities and prior to product transfer to other manufacturing operations.
The solution is within the organization. Objectives include: defining a continuum of drivers and variables, embedding process understanding, building an early warning and signaling process, and developing and adjusting process know-how. Statistical tools must be integrated into the fabric of the manufacturing operation for addressing deviations and managing change control.
Problematic for many pharmaceutical operations is a lack of fundamental understanding of their processes and products. When a deviation occurs, this lack of understanding makes it difficult to determine the root cause of the deviation. The investigator has no baseline to examine for sources of variation or causal events. Lack of root cause determination for deviations is a common FDA citation.
Organizing For Success
An increase in the use of statistics is inevitable. There is no single statistician that can possibly maintain all of the required skills at a high enough level to meet the overwhelming challenges of the new process validation guidance. There are alternative ways to address this challenge:
- Insource statistical skills
- Outsource statistical skills
- Hybrid approach
Insourcing completely requires a large staff, however complete coverage of statistical needs can be achieved. This is appropriate for large firms that can leverage these skills. However, the disadvantage is in the cost of maintaining this staff. Even large firms may face pressure to reduce the cost of analytics and may seek some level of outsourcing. In addition, a “next generation” gap could be forming as statisticians face retirement and replacements are strictly from academia without industry knowledge.
Outsourcing completely is appropriate for very small firms at the startup stage. However, this approach creates a dependency that may put the firm at continuity risk.
A Hybrid approach leverages the statistical competence of the existing internal staff while reaching out to fill the remaining skill gaps. This begins with the coupling of basic statistical training for the internal staff with a “back-room” network of external subject matter experts who supplement on project work as needed.
Conclusion
The use of statistical tools has the potential to greatly improve the process of demonstrating that scientific evidence exists to show that a process is capable of consistently delivering quality product, as required by the FDA Guidance. This more scientific approach also accelerates product development cycle-time and reduces the cost of regulatory compliance.
References
1. “Process Validation: General Principals and Practices” FDA Guidance
____________________________________________________________________________________
Merton Partners is a boutique-consulting firm that integrates a network of subject matter experts to meet the statistical needs of small to midsize pharmaceutical manufacturing operations and R&D organizations. Since 2008 Merton Partners has offered statistical services to drug development, process development, regulatory, CMC, and pharmaceutical manufacturing. We specialize in GMP compliance, validation and continuous improvement consulting and training. Contact Eugene Wordehoff at wordehoff@mertonpartners.com.